Cell Cluster Identification and Validation in Breast Tumor Tissue

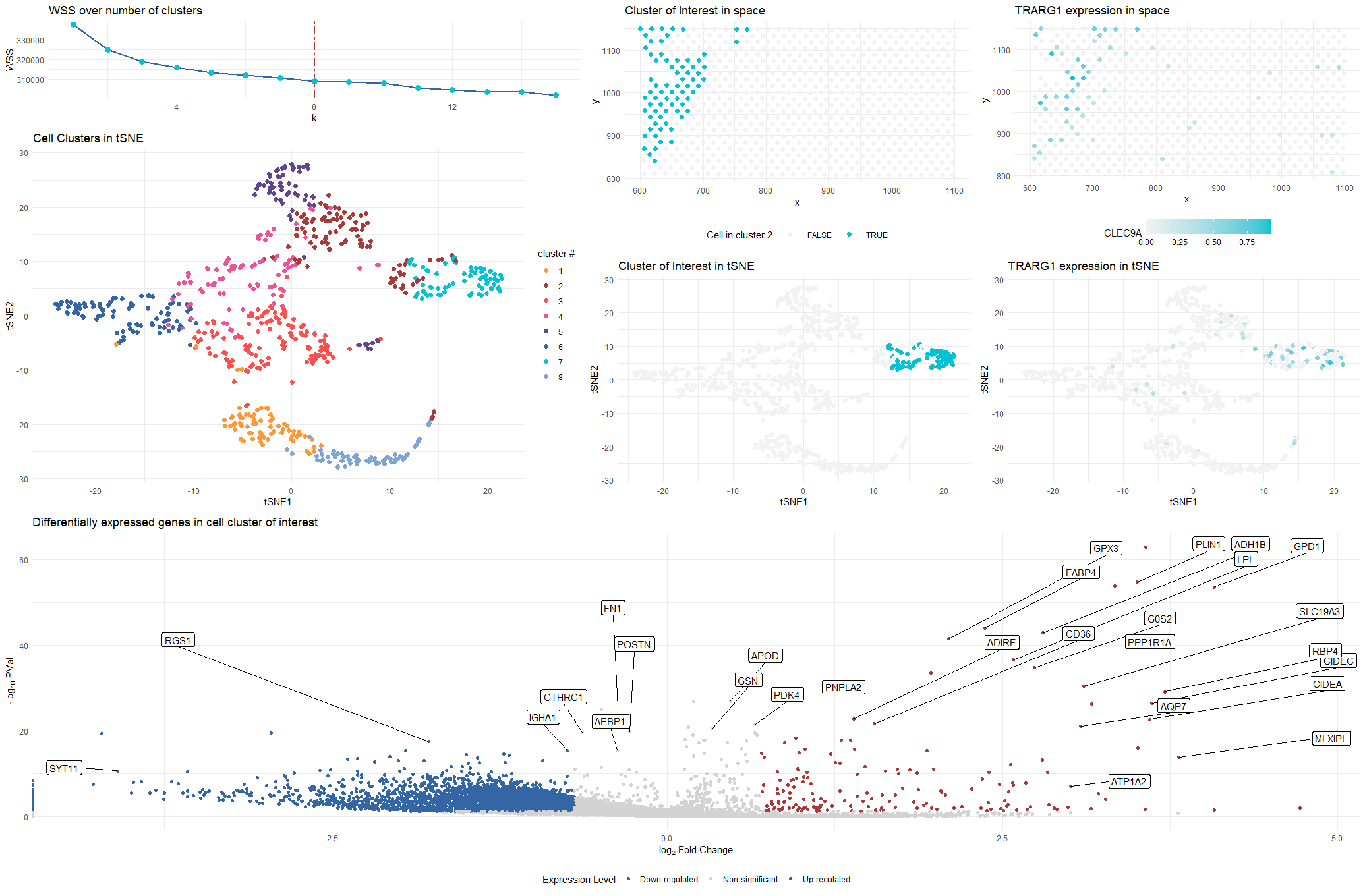
### Plot Description This visualization presents differential gene expression to validate cell type identification by k-means on 2D tSNE space.
-
The spatial-transcriptomics data on breast tumor tissue is preprocessed by removing cells with total gene counts below 0.2* average of total gene counts, normalized by total gene counts, and log-scaled.
-
The 8 cell clusters (see WSS curve for choice of k) are encoded by different colors in tSNE space to show the performance of k-means clustering.
-
A cluster is selected as cluster of interest by TARG1 count and identified in both tSNE and physical space with the same color used in the 8-cluster plot.
-
The most differentially expressed genes are computed in the selected cluster through two-sided Wilcox test, and demonstrated on the volcano plot.
-
The chracteristic TRARG1 gene’s distribution is plotted in both tSNE and physical space
Changes made
- Adapt to the columns of eevee dataset
- Change the number of PCs and Clusters based of scree plot and WSS
- Deal with NaN value in pvs
- attempt to use a more unique marker for human dendritic cells, so change the target cluster to Adipocytes (TRARG1)
- change the color as directed by TA feedback from HW4
ref: https://www.proteinatlas.org/ENSG00000184811-TRARG1
Source Code
## load dataset
data <- read.csv('C:/Users/ivych/OneDrive - Johns Hopkins/Classes/Data Visualization/eevee.csv.gz', row.names = 1)
data[1:5,1:5]
# install.packages("Rtsne")
## import libraries
library(ggplot2)
library(gridExtra)
library(Rtsne)
## set theme
#ref: https://www.color-hex.com/color-palette/1022322
xiao_plt <- c(
"#FF983B",
"#AA3A39",
"#FE4E4E",
"#EE5397",
"#634490",
"#3466A5",
"#00C4D4",
"#7fa4d6")
## data selection
pos <- data[, 2:3]
gexp <- data[, 4:ncol(data)]
gexp_mean <- mean(rowSums(gexp))
good.cells <-rownames(gexp)[rowSums(gexp) > 0.2*gexp_mean]
nrow(pos[good.cells,])
nrow(pos)
pos <- pos[good.cells,]
gexp <- gexp[good.cells,]
## normalization
gexp_norm <- gexp*median(rowSums(gexp))/rowSums(gexp)
gexp_log <- log10(gexp_norm+1)
mat <- unique(gexp_log)
#scree plot for pcs
x <- 1:15
pcs <- prcomp(mat)
par(mfrow=c(1,1))
data_scree <- data.frame(x = x, std = pcs$sdev[1:15])
ggplot(data_scree, aes(x, std)) +
geom_line(color = xiao_plt[4], size = 1) +
geom_point(color = xiao_plt[3], size = 3) +
labs(title = "Scree plot", x = "Number of PCs", y = "STD") +
theme_minimal()
#pcs = 4
#pcs and tsne
pcs_result <- pcs$x[,1:4]
tsne_result <- Rtsne::Rtsne(pcs_result)
## elbow method for clustering
wss <- numeric(15)
for (i in 1:15) {
wss[i] <- sum(kmeans(mat, centers = i)$withinss)
}
data_elbow <- data.frame(x = x, wss = wss)
p_elbow <- ggplot(data_elbow, aes(x, wss)) +
geom_segment(col = xiao_plt[2], size=1, linetype = 'twodash',
aes(x = 8, y = Inf, xend = 8, yend = -Inf)) +
geom_line(color = xiao_plt[6], size = 1) +
geom_point(color = xiao_plt[7], size = 3) +
labs(title = "WSS over number of clusters", x = "k", y = "WSS") +
theme_minimal()
p_elbow
## K-means
#k = 8 seems to give the optimal result
com = kmeans(mat, center = 8)
df <- data.frame(pos, tsne_result$Y, celltype=as.factor(com$cluster))
head(df)
p_k <- ggplot(df) +
geom_point(size = 2,aes(x = X1, y = X2, col=celltype)) +
labs(title = 'Cell Clusters in tSNE',
x = 'tSNE1', y = 'tSNE2', col = "cluster #") +
theme_minimal() + # theme(legend.position = "none") +
scale_colour_manual(values = xiao_plt)
p_k
# pick cluster of interest
coi <- 7
#cluster of interest in the tsne space
p_c1 <- ggplot(df) +
geom_point(size = 2,aes(x = X1, y = X2, col=(celltype==coi))) +
labs(title = 'Cluster of Interest in tSNE',
x = 'tSNE1', y = 'tSNE2',col="in cluster 2") +
theme_minimal() + theme(legend.position = "none") +
scale_colour_manual(values = c("grey95",xiao_plt[coi]))
p_c1
#cluster of interest in the physical space
p_c2 <- ggplot(df) +
geom_point(size = 2, aes(x = aligned_x, y = aligned_y, col=(celltype==coi))) +
labs(title = 'Cluster of Interest in space',
x = 'x', y = 'y',col="Cell in cluster 2") +
theme_minimal() + theme(legend.position = "bottom", legend.key.size = unit(1, "cm")) +
scale_colour_manual(values = c("grey95",xiao_plt[coi]))
p_c2
## Double-sided wilcox test to identify differential expression
cluster.oi<- names(which(com$cluster == coi))
cluster.ot <- names(which(com$cluster != coi))
#differential genes
genes <- colnames(mat)
pvs <- sapply(genes, function(g) {
oi <- mat[cluster.oi, g]
ot <- mat[cluster.ot, g]
wilcox.test(oi, ot, alternative="two.sided")$p.val})
pvs[is.na(pvs)] = 0.06
names(which(pvs < 1e-8))
head(sort(pvs), n=20)
#fold change
log2fc <- sapply(genes, function(g) {
oi <- mat[cluster.oi, g]
ot <- mat[cluster.ot, g]
log2(mean(oi)/mean(ot))})
log2fc[is.na(log2fc)] = 0
#volcano plot
df_volcano <- data.frame(pvs, log2fc)
exp_sig <- apply(df_volcano, 1, function(g) {
if (g[2] >= log(2) & g[1] <= 0.05) {out = "Up-regulated"}
else if (g[2] <= -log(2) & g[1] <= 0.05) {out = "Down-regulated"}
# else if (g[2] == 0 & g[1] == 0.06) {out = "NaN"}
else {out = "Non-significant"}
out})
data_deg <- cbind(df_volcano, exp_sig)
#install.packages("ggrepel")
p_volc <- ggplot(data_deg, aes(y=-log10(pvs), x=log2fc)) +
scale_color_manual(values = c(xiao_plt[6], "lightgray", xiao_plt[2])) +
geom_point(aes(col = exp_sig)) +
ggrepel::geom_label_repel(label=rownames(data_deg)) +
xlab(expression("log"[2]*" Fold Change")) +
ylab(expression("-log"[10]*" PVal")) +
theme_minimal()+
theme(legend.position = 'bottom') +
labs(title = 'Differentially expressed genes in cell cluster of interest',
col = "Expression Level")
p_volc
# Visualize CD1C
# https://bmccancer.biomedcentral.com/articles/10.1186/s12885-023-10558-2#:~:text=CD1C%20is%20an%20important%20part%20of%20the%20TME,and%20a%20new%20treatment%20target%20for%20breast%20cancer.
df_clec9a <- cbind(df,CLEC9A = mat$TRARG1)
p_clec9a <-ggplot(df_clec9a) +
geom_point(size = 2,aes(x = aligned_x, y = aligned_y, col = CLEC9A)) +
theme_minimal() + theme(legend.position="bottom", legend.key.width = unit(1, "cm")) +
labs(title = 'TRARG1 expression in space',
x = 'x', y = 'y') +
scale_color_gradient(low = 'grey95', high=xiao_plt[coi])
p_clec9a
p_clec9a2 <- ggplot(df_clec9a) +
geom_point(size = 2,aes(x = X1, y = X2, col = CLEC9A)) +
theme_minimal()+ theme(legend.position="none", legend.key.size = unit(0.2, "cm")) +
labs(title = 'TRARG1 expression in tSNE',
x = 'tSNE1', y = 'tSNE2') +
scale_color_gradient(low = 'grey95', high=xiao_plt[coi])
p_clec9a2
#plot panel arrangement
layout <- rbind(c(1,1,1,3,3,4,4),
c(2,2,2,3,3,4,4),
c(2,2,2,5,5,6,6),
c(2,2,2,5,5,6,6),
c(7,7,7,7,7,7,7),
c(7,7,7,7,7,7,7),
c(7,7,7,7,7,7,7))
grid.arrange(p_elbow,p_k,
p_c2,p_clec9a,
p_c1,p_clec9a2,
p_volc,
layout_matrix = layout)